
Hi everyone! Hope you’re ready for another weekly update from the Dorris Neuroscience Center at The Scripps Research Center in La Jolla! For those who don’t know what I’m doing, I am interning under my mentor Cailynn Wang, a research technician, and her work with CATCH (clearing-assisted tissue click chemistry). This week mainly focused on her HCR brain experiment from last week with the goal of determining whether the knockout virus injected into the mice brains actually worked by identifying the number of mRNA signals (from the NMDA receptors) present.
On Monday I stained the HCR slides with a DAPI stain, which highlights the nuclei of cells under a microscope. In short, HCR is the process of amplifying DNA or RNA signals so they can be identified through microscopy. After the HCR brains had been on the shaker with the DAPI stain for 15 minutes, I removed the brains and mounted them on slides for Cailynn to image later. I also spent some time talking to one of the MD/PhD students who is on a month-long rotation in the Ye Lab about what it means to be in the MD/PhD program and all the aspects of applying to medical school. I previously had no idea that such a program existed that allows one to obtain a PhD and medical degree in only 8 years and I think this is definitely and option I will consider in my future education. In the afternoon, Cailynn and I imaged half of the 24-brain sample we previously collected and cleared. These brains were used in a time-dependent experiment to determine the effects of increasingly longer amounts of time on sertraline retention in the brain. While imaging, we noticed that the drug signal was significantly less in the 4 and 8 hour mice than in the 1 hour mice, although no conclusions could be definitively drawn until the images were quantified on the computer. After that, we began imaging the HCR brains I mounted. Half of the brains were injected with an active virus that is supposed to decrease the number of NMDA receptors present in the brain, therefore reducing the drug retention because it has less areas to bind to. However, Cailynn has been struggling with achieving an effective virus and we were very anxious to see whether this new virus cocktail would be successful. After imaging under the confocal microscope, she was unsure whether there was a significant difference between the brain hemispheres treated with the active virus and those treated with the inactive control virus. Although this was not the result we were hoping for, it was progress because there did appear to be a slight decrease in the number of NMDA receptors present. At the end of the day I finished by making another batch of CLICK reaction buffer and transferring the other half of the 24-brain sample (which had been cleared in SDS for an addition 3 days in order to improve their transparency).
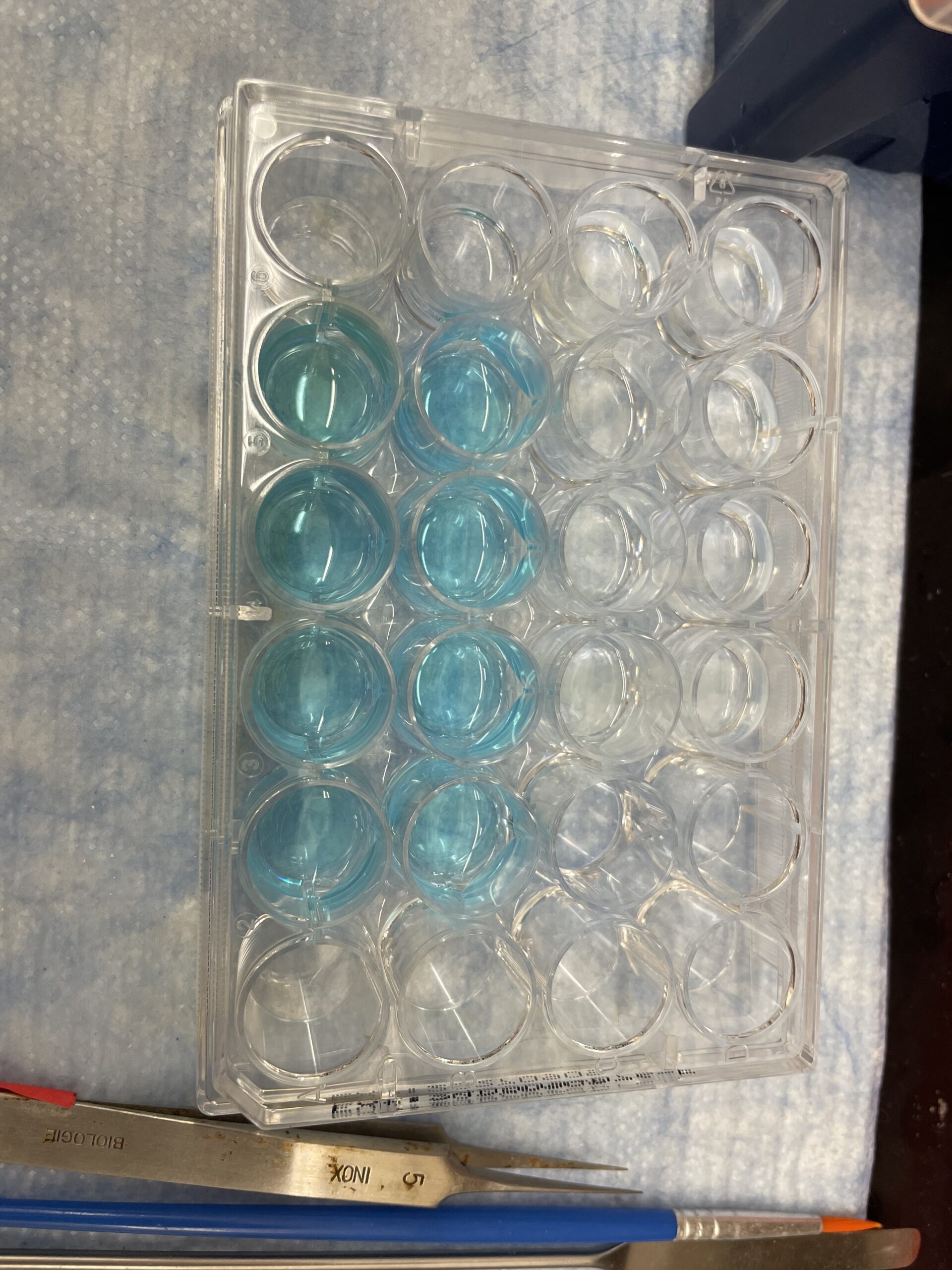
Running a CLICK reaction. The buffer is blue because of the presence of copper II that will be converted to copper I.
Tuesday began by changing the tissue I CLICKed yesterday into a buffer of PBST-EDTA. The tissue needs to be washed 3 times with the PBST-EDTA and then stained with DAPI before it can be mounted for imaging. Transferring the tissue from the CLICK buffer to the initial PBST-EDTA is a VERY tedious and time-consuming task because I had to label 24 new test tubes and meticulously transfer the delicate brain tissue from one tube to the next. This task took me the entire morning, and while I was busy changing buffers, Cailynn spent her morning analyzing the data from yesterday’s HCR imaging and concluded that there wasn’t much of a difference between the control virus and the active, knockout virus. After lunch I had a very cool opportunity to watch one of the postdocs, Ben, perform a xenograft (which is where he injects a tumor into the mice)! The mice Ben uses are immunocompromised (because otherwise the mice’s T-cells would fight off the injected tumor) so they are hairless and must be handled in a sterile area. Ben used a sub-cutaneous injection method to inject the tumor, meaning he placed the needle directly under the mouse’s skin. The purpose of his experiment is to identify the drug-tumor interaction on a single-cell level rather than on a broad, whole-system level that is typical in cancer studies. This is because most cancer treatments are effective against about 95% of tumor cells but fail to eliminate 100% of them, which leads to high rates of recurrence and tumor regrowth over time. This happens because tumors are composed of all different types of cells that all need to be studied individually while interacting with the drug in order to create truly effective treatments. After this, I returned to the lab (Ben’s injections took place in the vivarium) and checked in with Cailynn. She was analyzing more of the HCR brains (this time the virus was injected into the hippocampus rather than the cortex). Similar to the other HCR brains, there appeared to be minimal (yet still some) reduction in the number of NMDA receptors present. I finished the day by mounting 2 more slides before heading out.
Cailynn was running behind Wednesday morning so I began the day by researching how the confocal microscope works. Essentially, a confocal microscope works by focusing a highly specific laser on a single point in the tissue and then detecting the light emitted from that single point through a pinhole optically aligned with laser focus. This method significantly reduces the background/out-of-focus light emitted from standard microscopes, and therefore produces much higher-resolution images. These images are then combined on the computer to create 3D renderings of the tissues in a process called optical sectioning. Furthermore, confocal microscopy can identify multiple different cell components in one image by using fluorophores and various wavelengths of light. Specific cell structures are first labelled with fluorescent molecule stains (like DAPI for the nucleus). Each fluorophore has a unique absorption spectrum (wavelengths of light absorbed) and emission spectrum(wavelengths of light emitted). The laser on the confocal microscope can be fine-tuned to a specific wavelength of light that it emits, which enables scientists to select the appropriate excitation wavelength for chosen fluorophores (which will then only reveal those fluorophores). For example, the wavelength of light that shows the DAPI stain (and the cell nuclei) is 405 nm whereas the wavelength that illuminates the location of the drug is 647 nm. After Cailynn arrived around 11 I ran another CLICK reaction, but first had to determine the molarity of the sodium ascorbate to figure out how much PBS to add in order to create a properly-balanced solution. After lunch I mounted some of Cailynn’s lung tissue on slides which was frustratingly difficult today because I used to be able to do slides easily but today I couldn’t add a cover slide without getting an air bubble in it. Luckily the air bubble didn’t cover the tissue though so it wouldn’t effect the imaging quality. Cailynn and I then imaged these slides with the confocal. Cailynn’s previous lung samples had shown unknown “stars” in the tissue, which she wasn’t sure what they were, and we were hoping these lung samples would be clear. Unfortunately, the unknown “stars” still appeared on the new samples and Cailynn remained puzzled over their identity.

Mounting the lung tissue onto slides for imaging.
Thursday was my last day in the lab this week because of the holiday on Friday, and I began by doing some SAT practice in the morning as Cailynn was busy preparing for her chem-bio subgroup meeting later. Every 2 weeks in the Ye Lab there are subgroup meetings for the 3 subgroups (molecular bio, behavior, and chem-bio) that are supposed to allow members to present their current experiments or findings and get help from peers or advice on next steps. There are about 8 people in the chem-bio subgroup (which is a lot) but it means there are lots of ideas and advice for improving experiments. Cailynn’s presentation mainly focused on her recent HCR experiment and trouble-shooting the knockout (KO) virus that didn’t work so well. She also presented about the time-dependent experiment and its mixed results. The PI and a few postdocs offered some steps to try next, including injection with a covalent (rather than the non-covalent sertraline drug) in order to determine whether the perfusion itself caused the drug signals to move around or not show up under imaging.
Friday we went to Mission Beach, which wasn’t as crowded as I had expected, and enjoyed our holiday by soaking in the sun, making friendship bracelets, and boogie boarding in the waves. It felt good to be in the hot sun after a pretty cloudy and a little bit of a chilly week. Later, we came home and grilled burgers with the help of our house mom Jennie. We made s’mores and watched the fireworks before going to bed.
I woke up early Saturday and drove down to Mission Bay to get in a run before the sun heated up too much, then Maiya, Shiloh, Azari, and I spent a few hours at the mall before returning home and chilling with some reading and ping-pong. Sunday began with another run by the bay and then a trip to a local coffee shop with Maiya. I got a vanilla matcha which was delicious and very refreshing after my hot run.
I can’t believe I’ve already been at the lab for a month! I’m definitely getting much more comfortable here and I can’t believe I only have two more weeks to enjoy it. I am learning so much and gaining so much valuable experience in the real world. Can’t wait for week 5!
Thanks for making an effort to “dumb down” your scientific descriptions! What a great opportunity to chat with others in the lab.