
Hello everyone!! I’m Azari and I just finished week three at the Burton Lab at Scripps Research. Just a refresher, I’m working with Nate Beutler, a post-doctorate immunologist who, through antibody discovery and therapeutic design, is working to understand why an effective vaccine for malaria hasn’t been developed yet. This week I’ve slowly taken on more responsibilities and independence in the lab. The main highlights are transfecting our cells, purifying antibodies, and continuing to make great experiences here in Southern California.
Monday was very laid back. It was cell-splitting day so I calculated and split my cells on my own. Over the past few splits, I’ve grown very confident in my ability to do it independently so this only took about 10 minutes. Sean showed us the transfections from last week and unfortunately ours didn’t have much growth. I’m not too surprised because it is a very meticulous procedure and things could go wrong at any step. Sean’s transfection was successful and ready to be purified. He also showed us the BLI (biolayer interferometry) technique. BLI is used to study how a specific molecule (HIV antibody in Sean’s case) interacts with another (antigen). It measures how well an antibody sticks to an antigen. All of my mentors were busy with personal work meaning they didn’t have any work for us. Between meetings, Nate B. took us to Raising Canes which I was so happy about. He’s definitely gone up on my list of favorite people in the lab. Majority of the day Shiloh and I had time to work on personal things. I spent that time doing college prep so it was still a productive day!
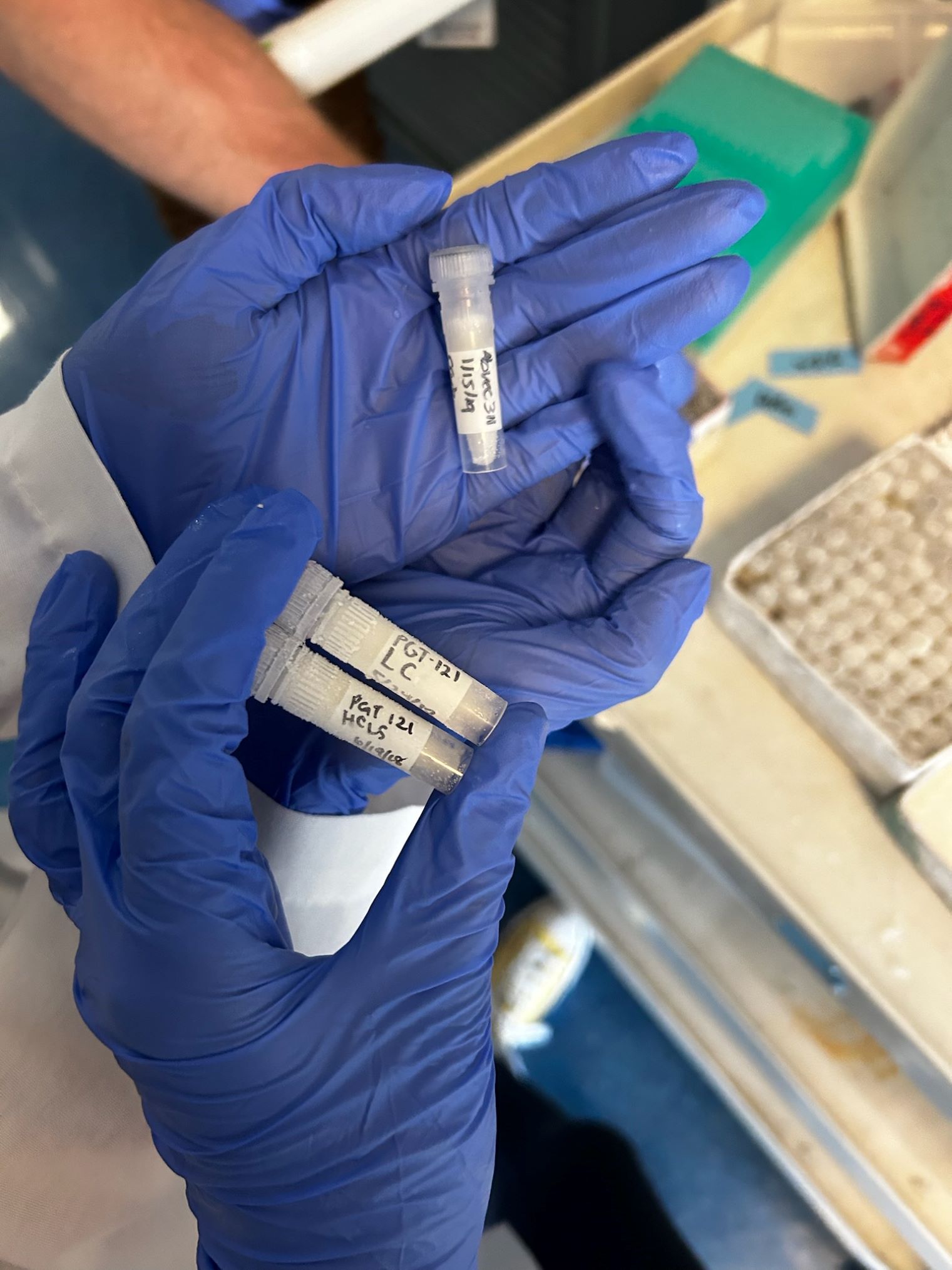
The antibodies we chose to grow
Tuesday was definitely more busy. Our cells were a day away from being ready to be transfected. To prepare, we needed to grow the DNA (antibody heavy chain and light chain) that we will be ‘infecting’ our cells with. First, we chose the antibodies we wanted to culture. I chose mAb311 (monoclonal antibody 311), which has strong inhibiting effects on malaria parasites. Shiloh chose a monoclonal HIV antibody since her mentors work more closely with that virus. We each had four samples: two sets of heavy chain and light chain of our antibodies, one set is resistant to carbonate and the other is resistant to kanamycin. Our samples were placed in the incubator overnight. Nate had to run over to UCSD to pick up some Plasmodium falciparum sporozoites and after asking profusely, he agreed to dropping Shiloh and I off at the Panda Express on campus (thanks Nate!!). When we returned, Yenni showed us another way of running proteins through a gel. The procedure was identical to how it’s usually done, just with a different dye that can be observed with the naked eye rather than under a UV light.

Our own boxes to put our antibody samples in!
Transfection day was certainly the most engaging. Our cells were at a great count to be transfected. Following the protocol, we calculated how much of each component was necessary. For mine, I needed 3ml of OptiMEM (enhances efficiency), 12.3ul of heavy chain, 20.88ul of light chain, 24ul of FectoPro (transfection reagent), and 27ml of cells. In one tube we combined our DNA and OptiMEM. It was incubated for 10 minutes before adding our cells and FectoPro. After its all mixed together its placed into the incubator. We took a small sample of our transfected solution to track how much of the antibody is expressed everyday. We will measure the samples later on. It may not sound like a lot but this procedure took us all day to complete. Between steps of our transfection, Yenni had us harvest his bacteria colonies from a petri dish to grow. He then had us run his DNA samples along with Shiloh and I’s heavy chain and light chain samples. Yenni also showed us our gel from yesterday and it looked great!
Expi293 cells need to be fed 24 hours after transfection. We fed them .3ml of glucose and .3ml of VPA and placed them back into the incubator to make the cells happy. Nate also showed us how to purify antibodies. The transfection solution is spun down and whatever is left is the supernatant. This supernatant contains our antibodies. Protein G sepharose (a kind of bead) is added; the antibodies stick to it. We want the antibodies on their own so we run them through a filter and rinse them off with PBS. Then we have them sit in 15 ml of .2m citric acid so the antibody can detach itself from the beads. It can’t sit for too long otherwise the antibody will fall apart. The acid-antibody solution is drained into a tube with 4.5ml of 2m tribase to neutralize the acid. A mixture of 3600ml of water and 400ml of PBS is created for the new solution to sit in. We put the solution in a permeable membrane (only allows things smaller than 10,000 daltons through) container. This allows the PBS in to keep the antibodies at a stable pH level. This has to sit overnight in the cold room. After Nate left, Shiloh and I checked the viability of our transfected cells. Mine were pretty low. We took another sample of our transfected cells to be measured in the future. Yenni showed us the gel he had us run yesterday and as it turns out we accidentally ran the wrong sample. He wasn’t upset, I actually think he found it funny. The next morning we finished purifying our antibodies. We took them out of the PBS solution they were sitting in all night and counted them. Our antibodies were way too concentrated so we diluted them before running them through a gel. The rest of the day was spent doing 16 minipreps for Sean. I’m really grateful that he trusted us enough to do it for him. He would’ve had to start over if we had messed up and luckily we didn’t!! Nate went with us to this concert hosted by Scripps Research which was really cool to see.
The Weeknd concert!!!
I’m halfway through my internship and experiences so far have been memorable and overall very meaningful to me. I’ve grown really confident in the lab which is something I was definitely nervous about before coming here. Everyone in the lab is very welcoming and open to letting me in on their work, even those who aren’t my official mentors. I’ve made amazing connections and experiences in and out of the lab that I will absolutely think back on in the future. This past weekend was definitely my favorite so far. All of us went to see The Weeknd in Los Angeles. If you didn’t know already, I’m a die hard fan of The Weeknd so this was insane to get to see. I got to go with the coolest girls ever which made it that much more fun. (Don’t ask how much I spent at this concert!!) I really look forward to what the next few weeks will look like, but I will certainly be sad when it’s all over.
Thanks for reading, see you next week : )
yay!!! this week was so awesome, I fear we are too cool 😀